User Guide#
The user guide will talk about how the pipeline is implemented and how to modified pipeline or setup a custom pipeline.
Overview of MATTE#

Gene clustering process#
Broadly speaking, there are three steps:
preprocessing
clustering
Module analysis
The results that each of these parameters can lead to and produce will be described in detail in the following three procedures.
Pipeline design#
PipeFunc is a most basic class in MATTE python package. The class
store a function and it’s arguments but not run it until calling it
like calling a funtion. And a Pipefunc will show it’s arguments and
name by print(PipeFunc) or str(PipeFunc). The string will be
updated everytime parameters are added by add_params, but not when
being called. When you call it, stored arguments can be ellipsised. But
arguments you put in will also be considered.
In a AlignPipe class, there are several PipeFunc and
transformer. Here, transformer means object that has fit and
transform methods. When calling AlignPipe.calculate,
PipeFunc and transformer will be performed one by one. All
funcs are stored in AlignPipe.funcs attributes and
AlignPipe.cluster_func attributes. One can use build-in function
add_steps to add a preprocessing step into AlignPipe.funcs
and set_cluster_method to add clustering step into
AlignPipe.cluster_func. These two are list object that store
PipeFunc and transformer. You can also add parameters by
add_param, (in this step, a new function that return a dict contain
param value and name will be generated) add transformer by
add_transformer. When you create a AlignPipe object, by setting
init=True, default pipeline will be created. All functions in the
pipeline should return a dict object,(all these returns are saved in a
dict temp_result) and by this way its return will be record and use
in the following functions.
There are four methods to run pipeline, calculate and
fit_transform is the same, but fit_transform has a parameters
that allow save temp result. The saved temp dict object can be used in
calculate_from_temp, where only clustering functions will be
performed. And transform will not fit the data but use fitted
transformers in the pipeline. If you want to use attributes of
transformers, use get_attribute_from_transformer.
Some key temps are following: * When running the pipeline, before
clustering there should generate a object named “before_clustering_df”
to used in the cluster_fun. * And in the preprocessing steps,
return object named “df_exp” to corver input. * When using
CrossCluster object to perform clustering, cluster_label and
cluster_properties will be passed to build_results * And
build_results will return a object named Result.
For convenience, a decorator utils.kw_decorator is used in most of
functions in pipeline. It makes a fucntion return a dict according to
parameter kw(str or list), and the function will accept
additional parameters but not raise error.
Preprocessing#
Key function in preprocessing is Kernel_transform, who perform
mostly important relative differential expression. Some kernel transform
is implemented: ‘meanrbf’,‘meanlocalrbf’,‘mean’,‘median’,‘medianrbf’ and
‘medianlocalrbf’. For normal, mean kernels is recommended, but with
outliers median function may have a better performance. rbf related
kernels performs a variant Gauss kernel transformation after RDE, and in
the localrbf kernel, each genes’ gamma is depended on it’s nearest K
neibors’ distance. A function is also accept to kernel_type
parameter. And this function should be performed in two vector, (it
should be acctepted by scipy.spatial.distance.cdist). But if the
funtion is of great complexities, it can cost much time for this step
will performed to each gene in the each phenotype. After kernel matrix
calculation, a double_centering and
sklearn.preprocessing.KernelCenterer will be performed by default.
In the previous test, these two will make results get more score, but
may change in your case.
Other functions’ detail can be seen in API page, exp_filter filter
out genes with extremely low expression(custom function is allowed too),
and RPKM2TPM is an important normalization preprocessing steps.
Keeping default preprocessing steps is highly recommended.
Clustering#
CrossCluster is clustering function or clusterer warpper. There are three pre-implemented methods: kmeans (wrapper from Bio.Cluster.kclust), spectrum and spectral_bicluster (wrapper from sklearn implemention). So other keywords parameters should seen in the original implemention. In spectrum clustering, by default will calculate distance matrix first(parameter use_aff). If not using preset implemention, by build_from_func or build_from_model to set up clustering methods.
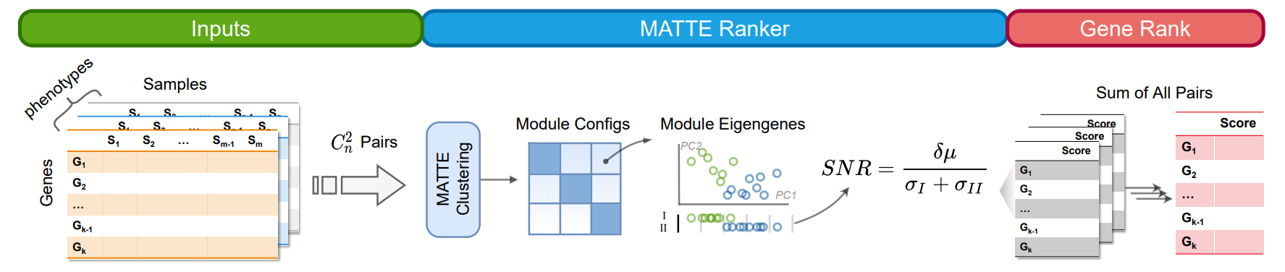
Gene Ranker#
GeneRanker accept more than two phenotypes, and will use pipeline to calculate pairs of phenotypes one by one, fincally sum/concatenate the results. Inputs of ModuleEmbedder is not the same as pipeline. Row of Expression data is sample, column is gene.
There are two types of ranking :
There are several types of GeneRanker: 1. ‘module’ and ‘gene’
will cluster genes according to their expression. And Use module SNR to rank genes. In gene mode, the SNR will be corrected by the correlation of gene expression and module eigen.
‘dist’,‘cross-dist’.
In dist mode, the distance of each genes will be calculated. And genes will be ranked according to the sum of distance to each other genes. In cross-dist mode, the distance of differential expression and differential co-expression will be merged.